
What is Fabry Disease?
Fabry disease (diffuse universal angiokeratoma, hereditary dystonic lipidosis) is a congenital disease characterized by a hereditary deficiency of the alpha-G4-galactosidase enzyme, resulting in the accumulation of glycolipids (ceramide) in the cytoplasm and lysosomes of cells of various organs and tissues.
It was described for the first time in 1898 by the English dermatologist Anderson and the German dermatologist Fabry.
Causes of Fabry Disease
Fabry disease is caused by a deficiency of lysosomal hydrolase – a-galactosidase A, inherited by the X-linked recessive type, and therefore the clinical symptoms occur in males. The a-galactosidase A gene is located on the long arm of the Xq22 q24 X chromosome.
Pathogenesis during Fabry Disease
An enzyme defect leads to systemic deposition of sphingoglycolipids and glycoproteins in the affected tissues, in particular in the endothelium and smooth muscles of the vessels, heart, kidneys (glomeruli and tubules), eyes (corneal epithelial cells), ganglia of the autonomic nervous system.
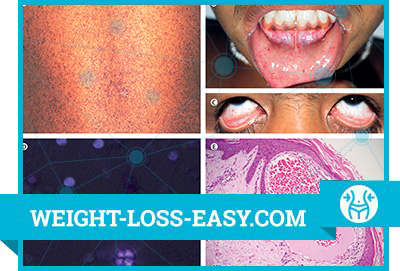
Symptoms of Fabry Disease
In homozygous men, the disease manifests itself in childhood or in adolescents with so-called Fabry crises, characterized by a painful burning pain in the palms and feet. Attacks of pain can last several days, accompanied by a slight fever and increased ESR. Sometimes rheumatism is mistakenly diagnosed. Acroparastesias become more frequent and more severe with age. In childhood, skin lesions characteristic of Fabry disease are noted – angiokeratomas (telangiectasias or small superficial angiomas), which are located symmetrically on the hips, back, buttocks, external genitals, in the area of the knee joints and navel. Angiokeratomas can be flat or somewhat elevated above the skin surface, from dark red to bluish-black color, they do not fade when pressed. Hypo-or anhidrosis of the skin is characteristic. It is also possible damage to the mucous membrane of the mouth and conjunctiva. Observed expansion and tortuosity of the retina and conjunctiva, clouding of the cornea. With age, the accumulation of sphingoglycolipids and glycoproteins in the organs of the cardiovascular system and kidneys increases. Symptoms of stenocardia appear, heart valves are affected (mitral insufficiency and aortic stenosis), left ventricular hypertrophy, myocardial infarction develop.
Observed proteinuria, reduced filtration ability of the kidneys. Chronic renal failure may develop. Arterial hypertension (presumably of renal origin) intensifies the disturbances of the heart and contributes to the defeat of the cerebral vessels. Cerebrovascular manifestations include thrombosis, aneurysms, sometimes hemorrhages, which leads to seizures, hemiplegia, aphasia; possible psychosis. Changes in the gastrointestinal tract are characterized by nausea, vomiting, diarrhea, pain in the abdomen. Deformities of the distal interphalangeal joints, aseptic necrosis of the femoral head and the talus bones, and marked vertebral osteoporosis are noted. Hypochromic microcytic anemia, growth retardation and puberty are possible. Death occurs from uremia or vascular lesions of the heart and brain, usually in the 4th decade of life.
In heterozygous women, the symptoms of the disease may be absent, in the majority of carriers of the Fabry disease gene, skin manifestations and corneal clouding are observed, sometimes the symptoms of the disease are expressed in the same way as in men.
Diagnosis of Fabry Disease
The diagnosis is based on clinical manifestations, detection of a-galactosidase A activity in plasma, leukocytes, lacrimal fluid, cultured fibroblasts, tissue biopsies, as well as an increase in sphingoglycolipids (in particular trihexosilceramide) in urine, plasma, cultured fibroblasts. The differential diagnosis is carried out with hereditary hemorrhagic telangaectasia, in which there are telangiectasias in different parts of the body, bleeding, but there are no attacks of pain, kidney and cardiovascular system, gastrointestinal tract characteristic of Fabry disease. Differential diagnosis with rheumatism is based on skin changes and differences in biochemical parameters characteristic of Fabry disease.
Fabry Disease Treatment
Diphenylhydantoin and carbamadepine, corticosteroids, are prescribed to relieve and prevent pain attacks. In some cases, a kidney transplant is indicated. Attempts are being made to introduce the purified enzyme, however, the half-life of the enzyme in the body is short.
Prevention of Fabry Disease
Prenatal diagnosis is possible, which consists in the study in cultured amniotic cells of a-galactosidase A activity. This is the most effective method of preventing the birth of a sick child.